Kroos and Yu Supplemental Information
Primers for QuickChange (Stratagene) Site-directed Mutagenesis of spoIVFB.
| Substitution | Primer | Sequence |
| E44Q | LK245 LK246 |
5' GATTGTATTGATTCATCAGCTGGGGCATGCTGC 3' a 5' GCAGCATGCCCCAGCTGATGAATCAATACAATC 3' |
| E44A | LK247 LK248 |
5' GATTGTATTGATTCATGCGCTGGGGCATGCTGC 3' 5' GCAGCATGCCCCAGCGCATGAATCAATACAATC 3' |
| E44D | LK249 LK250 |
5' GATTGTATTGATTCATGACCTGGGGCATGCTGC 3' 5' GCAGCATGCCCCAGGTCATGAATCAATACAATC 3' |
| I42P | LK251 LK252 |
5' GCTCCTGATTGTATTGCCTCATGAGCTGGGGC 3' 5' GCCCCAGCTCATGAGGCAATACAATCAGGAGC 3' |
| D137H | LK253 LK254 |
5' CGATCTGGCCGCTGCATGGAGGAAAACTG 3' 5' CAGTTTTCCTCCATGCAGCGGCCAGATCG 3' |
| D137N | LK255 LK256 |
5' CGATCTGGCCGCTGAATGGAGGAAAACTG 3' 5' CAGTTTTCCTCCATTCAGCGGCCAGATCG 3' |
a Underlining indicates the mutagenic nucleotide(s).
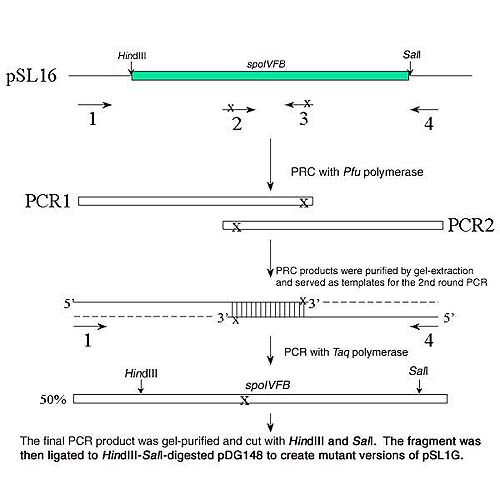
Site-directed mutagenesis of spoIVFB using sequence-overlap extension (SOE).
SOE employs 2 rounds of PCR to generate mutations in the desired position of the gene. The protocol was as follows: (see the diagram)
The first round of PCR: Plasmid pSL16 (100 ng ) was used as the template. Two external primers (flanking spoIVFB) and two internal primers (each containing a mutated nucleotide) were used to generate PCR products 1 and 2 in separate reactions. The primers were designed to create an overlap region in the two PCR fragments. To reduce the chance of making undesired mutations during the PCR, DNA polymerase Pfu ( Stratagene) was used in this step. The thermal cycling conditions were 94° C for 3 min; 30 cycles of 94° C for 1 min, 50°C for 1 min, and 72°C for 2 min; 72°C for 10 min. The first round PCR products were then purified by gel-extraction (Promega).
The second round of PCR: Purified PCR products 1 and 2 (100 ng each) served as templates. The two external primers were used to amplify the full-length SOE product. The above PCR conditions were modified slightly: Taq DNA polymerase was used for 25 cycles of amplification. Half of the PCR products are expected to contain the mutation in Primer 2 (see diagram) and the other half contain the mutation in Primer 3 (not shown in the diagram).
The final SOE products were cut with HindIII and SalI restriction enzymes, and the 1.0-kbp fragments containing the full-length spoIVFB gene were then ligated to HindIII-SalI-pDG148 to create mutant versions of pSL16.
| Substitution | Primer 1 | Primer 2 | Primer 3 | Primer 4 |
| D137A D137E H43F H47F |
LK152 LK152 LK152 LK152 |
LK140 LK160 LK178 LK180 |
LK161 LK161 LK179 LK181 |
LK153 LK153 LK153 LK153 |
| Primer | Sequence | Mutation |
| LK140 | 5’GGCCGCTGGCTGGAGGAAA3’ | SpoIVFBD137A |
| LK160 | 5’GGCCGCTGGAAGGAGGAAAA3’ | SpoIVFBD137E |
| LK178 | 5’TGTATTGATTTTTGAGCTGGGGCA3’ | SpoIVFBH43F |
| LK180 | 5’CATGAGCTGGGGTTTGCTGCTCTGGC3’ | SpoIVFBH47F |
| LK161 | 5’GAAAAGGCAGCTATTTGGAAAAC3’ | SpoIVFB1-148 |
| LK179 | 5’TGCCCCAGCTCAGCAATCAATACAAT3’ | SpoIVFBH43A |
| LK181 | 5’GCCAGAGCAGCAGCCCCCAGCTCATG3’ | SpoIVFBH47A |
| LK152 | 5’AGTGGTCAAGACCTCACTAG3’ | |
| LK153 | 5’CGGAATTAGCTTGCATGCCT3’ |
Generation of anti-SpoIVFB antiserum and affinity purification of antibodies.
A 292-bp DNA fragment generated by the polymerase chain reaction with pSC224 [Cutting, 1991 #484] as the template using upstream primer 5' GGCATATGGAGGAATATCGGCAAAGG 3' (NdeI site is underlined) and downstream primer 5' GCGGATCCTTAGTAGGGCAGAAGCAGTTCCTC 3' (BamHI site is underlined) was digested with NdeI and BamHI, and ligated to NdeI-BamHI-digested pET16b (Novagen), resulting in pSL28a, in which the last 92 amino acid residues of SpoIVFB are preceded by 10 histidines under the control of a T7 RNAP promoter. The fusion protein was expressed in E. coli BL21 lDE3/pLysS (Novagen) by adding IPTG (1 mM) when the culture reached the late log phase of growth and continuing incubation for 3 h at 37°C. Proteins were separated by SDS-PAGE, the His10-SpoIVFB197-288 chimera was sliced from the gel, and electroeluted protein was injected into rabbits to generate an antiserum.
To affinity purify anti-SpoIVFB antibodies, the His10-SpoIVFB197-288 fusion protein was purified and used to create an affinity column. Cells from a culture (150 ml) induced to produce the fusion protein as described above were resuspended in 5 ml buffer A (40 mM sodium phosphate pH 8.0, 6 M guanidine-HCl, 0.5 M NaCl) and stirred for 2 h at 25°C. The lysate was centrifuged at 12,000 g for 15 min at 4°C and the supernatant was mixed with 3 ml of a 50% suspension of Ni-NTA-agarose (Qiagen) in buffer A. The mixture was loaded into a column, which was then washed with 10 volumes of buffer B (40 mM sodium phosphate pH 8.0, 6 M urea, 0.5 M NaCl) followed by 10 volumes of buffer C (buffer B at pH 6.2), and proteins were eluted with buffer D (buffer B at pH 4.7). Eluted proteins were concentrated 4-fold by centrifuging in a Centrex UF-2 ultrafilter (Schleicher & Schuell) and dialyzed against buffer E (20 mM sodium phosphate pH 7.1, 3 M guanidine-HCl, 0.5 M NaCl). The fusion protein was coupled to Affigel-10 (Bio-Rad Laboratories) and a 20 ml column was used to affinity purify the polyclonal antiserum as described previously [Resnekov, 1996 #708] except the column was washed with at least 40 volumes of 10 mM Tris-HCl pH 7.5 and 40 volumes of the same buffer plus 0.5 M NaCl until no protein was detectable in the eluate by the Bradford assay [Bradford, 1976 #478]. Antibodies were eluted from the column using 0.2 M glycine, 1 mM EDTA pH 2.2 and immediately neutralized by adding 0.5 volume of 2 M Tris-HCl pH 8.5.
1. Cutting, S., S. Roels, and R. Losick. 1991. Sporulation operon spoIVF and the characterization of mutations that uncouple mother-cell from forespore gene expression in Bacillus subtilis. J. Mol. Biol. 221:1237-1256.
2. Resnekov, O., S. Alper, and R. Losick. 1996. Subcellular localization of proteins governing the proteolytic activation of a developmental transcription factor in Bacillus subtilis. Genes Cells 1:529-542.
3. Bradford, M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254.